| This webinar is part of a series run by the ELIXIR 3D-BioInfo Community. There is a complete list of webinars here. |
Hosts

Barcelona Supercomputing Center

University College London (UCL)
Programme
The encyclopaedia of domains
Dr Nicola Bordin (UCL – CATH Lab)

The Encyclopaedia of Domains (TED) is a comprehensive classification of all globular protein structure domains in AlphaFold Database v4. Harnessing state-of-the-art deep learning methods for domain detection, structure comparison and fold detection, TED segments and classifies domains across AFDB, identifying over 370 million distinct domains, surpassing sequence-based resources by over 100 million domains. Nearly 80% of these domains exhibit similarities with known superfamilies in CATH, expanding the resource by over 600-fold. The remaining 20% that do not have relatives in any PDB-based resources unveiled over 7 thousand new folds, some of which have interesting and beautiful symmetries.
We also find some fascinating new architectures. TED uncovers over 10,000 previously undetected structural interactions between superfamilies and extends domain coverage to over 1 million taxa, enhancing research for organisms which previously had low to non-existent structural coverage. TED data will be made available in 3D-Beacons as well as a dedicated resource, significantly enriching CATH superfamilies.
Towards a complete structural map of the human proteome
Prof. Arne Elofsson (Stockholm University)
Cellular functions are governed by molecular machines that assemble through protein-protein interactions. Their atomic details are critical to studying their molecular mechanisms. Today the structure of virtually all individual proteins is available from predictions using AlphaFold. However, these predictions are limited to individual chains and do not include interactions. In this talk I will describe our attempts to increase the structural coverage of protein-protein interactions. Today fewer than 5% of hundreds of thousands of human protein interactions have been structurally characterised.
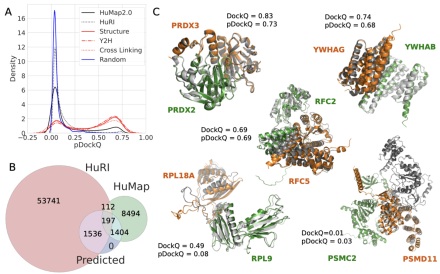
We show that combining predictions and experiments can orthogonally confirm higher-confidence models, and using AlphaFold2, we have built 3,137 high-confidence models, of which 1,371 have no homology to a known structure. We are exploring rapid methods to identify protein interaction networks. Finally, we show how the predicted binary complexes can be used to build very larger assemblies using a Monte Carlo Tree search method.
You can find previous webinars from the 3D-BioInfo Community on the Community webinars page.